Для термодинамического цикла интеграл по замкнутому контуру равен нулю
Поэтому подынтегральная функция, равная элементарной приведенной теплоте dQобр/Т, является полным дифференциалом dS, который Клаузиус назвал энтропией. Направление самопроизвольного процесса в изолированной системе связано с изменением энтропии dS.
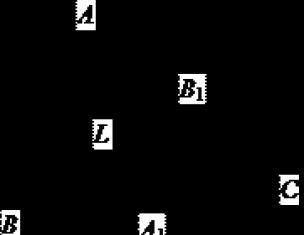
Второй закон термодинамики: в изолированных системах самопроизвольно могут совершаться только такие процессы, при которых энтропия системы Sизол возрастает, и процесс может идти самопроизвольно только для такого состояния, при котором энтропия обладает максимальным для данных условий значением Sмах. Рис. 1.7 является пояснением второго закона термодина-мики.
Некоторые другие формулировки второго закона термодинамики:
1. Никакая совокупность процессов не может сводиться только к превращению теплоты в работу, тогда как превращение работы в теплоту может быть единственным результатом процессов (Томсон).
2. Невозможно создание вечного двигателя второго рода, т.е. машины, которая производила бы работу только за счет поглощения теплоты из окружающей среды без передачи части теплоты холодильнику (Оствальд).
 |
Прямой процесс Равновесие Обратный процесс
Рис. 1.7. Изменение энтропии изолированной системы Sизол
в самопроизвольном прямом и обратном процессах согласно
второму закону термодинамики
Математическая формулировка второго закона термодинамики:
dSизол ≥ 0, ТdSизол ≥ dQ. (1.104)
Выразив теплоту на основании первого закона термодинамики через изменение внутренней энергии dU и произведенную работу dW, получим объединенное уравнение первого и второго закона термодинамики:
ТdS ≥ dU + dW или -dW ≥ dU – ТdS. (1.105)
Общий критерий направления самопроизвольного процесса, применимый как для изолированной, так и для закрытой, открытой систем, можно получить, применяя фундаментальное уравнение Гиббса. Из (1.32) с учетом второго закона получим объединенное уравнение первого и второго начала термодинамики в виде:
DWполезн. ≥ dU + рdV – ТdS. (1.106)
Так как при р = const, Т = const drG = dU + pdV – TdS, то в условиях равновесия, когда система не производит работу,
drG = -dWполезн., drG = 0, (1.107)
в самопроизвольном необратимом процессе, когда в системе совершается в ходе реакции положительная работа,
drG < -dWполезн., drG < 0. (1.108)
Можно показать аналогичным образом, что при V = const, Т = const критерием самопроизвольного процесса и равновесия служат соотношения:
dА < 0, самопроизвольный процесс; dА = 0, равновесие.
Характер изменения ΔrG и ΔrА в химических процессах приведен на рис. 1.8.
р, T = const V, T = const
B хравн D B хравн D
Рис. 1.8. Изменение энергии Гиббса G при р = const, Т = const и энергии Гельмгольца при V = const, Т = const в ходе реакции В↔D: 1 – состояние равновесия, ΔrG = 0 и ΔrА = 0; 2,3 – направление самопроизвольного процесса, ΔrG < 0 и
ΔrА < 0; 4,5 – направление несамопроизвольного процесса, который возможен только при воздействии внешней силы, ΔrG > 0 и ΔrА > 0; xD – молярная доля вещества D, хравн соответствует химическому составу системы при равновесии
Задача 1.20. Определить направление самопроизвольного процесса
МgСО3(т) = МgО(т) + СО(г), Т = 298 К,
если ΔrНо = 100,8 кДж/моль, ΔrSо = 175,7 Дж.моль-1.К-1, ΔrUо =
98,3 кДж/моль.
ΔrGо = ΔrНо – ТΔrSо = 100,8 – 298.0,1757 = 48 кДж/моль,
ΔrА = ΔrU – ТΔrSо = 98,3 – 298.0,1757 = 45,9 кДж/моль.
Расчеты проведены для прямого направления. Так как в прямом направлении ΔrG > 0 и ΔrА > 0, то при заданной температуре процесс самопроизвольно протекает в обратном направлении.
Задача 1.21. Вычислить величины ΔG и ΔA в процессе полиморфного превращения: Sромб → Sмон при 25 оС, если при этой температуре и давлении 1 атм значения энтропии ромбической и моноклинной серы равны 31,88 и 32,55 Дж/моль.К, а стандарт-
ные теплоты сгорания их равны соответственно -296813 и
297148 Дж/моль. В первом приближении можно пренебречь различием в плотностях обеих модификаций серы при этих условиях. Какой вывод можно сделать об устойчивости модификаций серы?
Фазовый переход при равновесии между фазами не сопровождается изменением энергии Гиббса. Однако если он происходит не в условиях равновесия, то ΔG ≠ 0 и по знаку при значении ΔG можно судить о том, в каком направлении он будет идти самопроизвольно. Вычислим значения ΔG и ΔА:
ΔG =ΔН – ТΔS = ΔсНоS ромб – ΔсНоS мон – 298(SSмон – SSромб),
ΔG = -296813 + 297148 – 298(32,55 – 31,88) = 335 – 200 =
115 Дж/моль,
ΔА = ΔG – рΔV ≈ ΔG = 115 Дж/моль.
Поскольку ΔG > 0 и ΔА > 0, самопроизвольно процесс пойдет справа налево. При 25 оС и 1 атм более устойчивой модификацией будет ромбическая сера.
1.2.7. Изменение энтропии в физико-химических процессах
Энтропия вещества S изменяется при нагревании и в фазовых переходах. При нагревании количества вещества n моль от Т1
а) если р = const, то ΔS = n∫ СрdТ/Т, (1.109)
б) если V = const, то ΔS = n∫ СVdТ/Т. (1.110)
Рассмотрим решение данных уравнений для ряда типовых процессов.
1. При нагревании идеального газа:
а) если Ср = а + вТ + сТ2, то
DS = n, (1.111)
если Ср = const, то DS = n[Срln(T2/T1) + Rln(p1/p2)]; (1.112)
в изобарном процессе р = сonst и поэтому последний член уравнения Rln(p1/p2) не учитывается;
б) если Сv = а + вТ + сТ2, то
DS = n, (1.113)
если Сv = const, то DS = n[Сvln(T2/T1) + Rln(V2/V1)]; (1.114)
в изохорном процессе V = сonst и поэтому последний член уравнения Rln(V2/V1) не учитывается;
в) в изотермическом процессе Т = const,
DS = nRln(V2/V1) = nRln(р1/р2). (1.115)
2. При нагревании твердого или жидкого вещества не учитываются в уравнениях изменения энтропии (1.111) – (1.114) последние члены уравнений Rln(р1/р2) и Rln(V2/V1).
3. В изотермических фазовых переходах:
а) р = const, DS = nDНфп/Тфп, (1.116)
б) v = const, DS = nDUфп/Тфп, (1.117)
где DНфп и DUфп – тепловые эффекты фазовых переходов, Тфп – температура фазового перехода.
Наибольший прирост энтропии наблюдается обычно в процессе кипения (испарения) жидкости, поэтому энтропия газов (паров) значительно выше энтропии жидких и твердых тел. Обычно при комнатной температуре SоТ ≈ 40 – 50 Дж/моль.К, Sож ≈
≈ 80 – 160 Дж/моль.К и Sог ≈ 120 – 240 Дж/моль.К. Правило Трутона: для жидкостей, не сильно ассоциированных, при нормальной температуре кипения (рпар = 1 атм) увеличение энтропии примерно одинаково для разных жидкостей в процессе кипения и равно:
DSкип = DНпар/Ткип = 88 – 92 Дж/моль.К.
4. При совместном нагревании вещества и переходе его из твердого состояния в жидкое, а затем в газообразное необходимо суммировать все изменения энтропии и теплового эффекта:
твердое жидкое газообразное
состояние состояние состояние
Т1 ¾¾¾® Тпл ¾¾¾® Ткип ¾¾¾® Т2
нагрев плав- нагрев испа- нагрев
ление рение
Тпл Ткип Т2
ΔSт = ∫СртвdТ/Т + DНпл/Тпл + ∫СржdТ/Т + DНисп/Ткип + ∫СргdТ/Т, (1.118)
Т1 Тпл Ткип
Тпл Ткип Т2
ΔНт = ∫СртвdТ + DНпл + ∫СржdТ + DНисп + ∫СргdТ. (1.119)
Т1 Тпл Ткип
Здесь ΔSт и ΔНт – суммы всех изменений энтропии и энтальпии на пути от температуры Т1 до Т2; Тпл и Ткип – температуры плавления и кипения; Сртв, Срж, Срг – теплоемкости вещества в твердом, жидком и газообразном состоянии; DНпл и DНисп – тепловые эффекты плавления и испарения.
5. Взаимная диффузия газов. При смешении газов энтропия системы изменяется, так как происходит диффузия, проникновение газов друг в друга. Пусть два идеальных газа количеством n1 и n2 занимали объем V1 и V2. При смешении газов общий объем V= V1 + V2. изменение энтропии для первого газа ΔS1 и для второго газа ΔS2 равно
DS1 = n1Rln(V/V1), DS2 = n2 Rln(V/V2). (1.120)
Общее изменение энтропии: DS = DS1 + ΔS2. Введем молярные доли
х1 = n1/(n1 + n2), х2 = n2/(n1 + n2).
Тогда DS = -R(х1lnх1 + х2lnх2). (1.121)
6. Процесс необратимой химической реакции проходит с участием нескольких веществ, исходных и продуктов. Поэтому в термодинамических уравнениях применяется суммарное изменение теплоемкости системы DСр, значение которого рассчитывается с помощью суммарных коэффициентов Dа, Dв, Dс и Dс’ (уравнение (1.95)). Для расчета изменения энтропии химической реакции можно применять уравнения (1.111) – (1.114), где используются коэффициенты Dа, Dв, Dс вместо а, в, с.
Изменение теплового эффекта реакции c температурой можно рассчитать по уравнению (1.100).
Задача 1.22. Один моль алюминия нагревают от 298 К до
873 К. Зависимость молярной теплоемкости кристаллического алюминия от температуры:
Ср = 20,945 + 10,73.10-3 Т, Дж.моль-1.К-1.
Вычислить изменение энтропии.
ΔS = ∫СрdТ/Т = n(аln(Т2/Т1) + в(Т2 – Т1)) =
20,945ln(873/298) + 10,73.10-3(873 - 298) =
28,68 Дж.моль-1К-1.
Задача 1.23. Камеру с 400 г водорода объемом V1 при Т =
298 К соединяют с камерой вакуума такого же объема. Вычислить изменение энтропии при расширении идеального газа.
Водород диффундирует в вакуум и в результате занимает объем V = 2V1. Молярная масса водорода 2 г/моль. Количество молей газа n = 400/2 = 200 моль. Изменение энтропии в процессе диффузии равно
DS = nRln(V/V1) = 200.8,31.ln2 = 1152 Дж.К-1.
Задача 1.24. Определить изменение энтропии в фазовом переходе при испарении 400 г диэтилового эфира (С2Н5)2О, давление стандартное; Ткип = 307,7 К; молярная теплота испарения DНисп = 27,2 кДж.
Молярная масса М и количество молей эфира n равны
М(С2Н5)2О = 74 г/моль, n = 400/74 = 5,4 моль.
Изменение энтропии
DS = nDНисп/Т = 5,4.27200/307,7 = 477 Дж.К-1.
Задача 1.25. Определить изменение энтропии при плавлении
1 моля льда, если Тпл = 273,2 К и DНпл = 6008 Дж/моль.
Применяем уравнение (1.116):
DSпл = DНпл/Тпл = 6008/273,2 = 22,0 Дж/моль.К.
Задача 1.26. Определить изменение энтропии при изотермическом расширении 1 моля идеального газа, если давление изменяется от 1 до 0,1 атм.
Для идеального газа при постоянной температуре V2/V1 =
DS = Rln(V2/V1) = Rln(р1/р2) = 8,314. ln(1/0,1) = 19,1 Дж/моль.К.
Задача 1.27. Определить изменение энтропии при изотермическом смешении различных идеальных газов, n1 = n2 = 1 моль.
Объем каждого из газов до смешения равнялся V, после смешения 2V. Изменение энтропии:
DS = Rln(2V/V) + Rln(2V/V) = 2Rln2 =
2.8,314.0,693 = 11,52 Дж/моль.К.
Задача 1.28. Вычислить изменение энтропии при неравновесном переходе в лед 1 моля воды, переохлажденной до -5 оС, если DНпл = 6008 Дж/моль.К при 0 оС.
Изменение энтропии в неравновесном процессе можно вычислить, заменяя его некоторой совокупностью равновесных процессов, происходящих между теми же начальными и конечными состояниями. Для расчета заменим неравновесный процесс:
1. Н2Ож(t = -5оС) → Н2От(t = -5оС), DS1 = ?
следующим сочетанием равновесных процессов:
2. Нагрев воды от -5 оС до 0 оС:
Н2Ож(t = -5 оС) ↔ Н2Ож(t = 0 оС).
Изменение энтропии равно
ΔS2 = ∫(Ср,ж/Т)dТ = 76,00ln(273,2/268,2) = 1,40 Дж/моль.К.
3. Равновесная кристаллизация воды при 0 оС
Н2Ож(t = 0 оС) ↔ Н2От (t = 0 оС),
DS3 = DНпл/Тпл = -6008/273,2 = -21,99 Дж/моль.К.
4. Охлаждение льда от 0 оС до -5 оС:
Н2От(t = 0 оС) ↔ Н2От (t = -5 оС),
ΔS4 = ∫Ср,т/ТdТ = 37,66ln(268,2/273,2) = -0,70 Дж/моль.К.
Цикл для расчета изменения энтропии в неравновесном процессе
Н2Ож, t = -5 оС Н2От, t = -5 оС
Н2Ож, t = 0 оС DS3 Н2От, t = 0 оС
Изменение энтропии в неравновесном процессе:
DS1 = DS2 + DS3 + DS4 = 1,4 – 21,9 – 0,7 = -21,29 Дж/моль.К.
Задача 1.29. Вычислить изменение ΔGо при изобарном нагреве 1 моля NH3 от 300 до 400 К при р = 1 атм, если стандартная энтропия аммиака Sо298 = 192,5 Дж/моль.К, а средняя теплоемкость в этом интервале температур Ср = 35,65 Дж/моль.К.
Чтобы определить изменение энергии Гиббса при нагреве вещества от температуры Т1 до температуры Т2, воспользуемся зависимостью ΔfGо от ΔƒНо и Sо:
ΔƒН0(Т2) – Δƒ Н0(Т1) = ∫СрdТ, S0Т2 = S0Т1 + DS,
T2SоT2 – T1SоT1 = (T2 – T1) SоT1 + DS,
ΔfGо(Т2) – ΔfGо(Т1) = ∫СрdТ – (Т2 – Т1)S0 T1 – Т2 ∫(Ср/Т)dТ.
В небольшом интервале температур, когда можно принять теплоемкости постоянными, уравнение упростится до вида
ΔfGо(Т2) – ΔfGо(Т1) = (Ср – S0T1)((Т2 – Т1) – Т2Срln(Т2/Т1).
ΔGо = (35,65 – 192,5)(400 – 300) – 400.35,65ln(400/300) =
17,47 кДж/моль.
Задача 1.30. Определить изменение энтропии DSТ и изменение теплового эффекта образования вещества DНТ при нагревании от 260 К до 430 К десяти килограммов уксусной кислоты, если плавление характеризуется температурой Тпл = 298,8 К и теплотой плавления DНпл = 11700 Дж/моль, кипение характеризуется температурной Ткип = 391,4 К и теплотой испарения DНисп =
24400 Дж/моль, а средняя удельная теплоемкость вещества в твердом, жидком и газообразном состояниях соответственно
равна Ĉр = 2039 Дж.кг-1.К-1, Ĉрж = 2057 Дж.кг-1.К-1, Ĉрг =
1197 Дж.кг-1.К-1.
Для вычисления DSТ и DНТ применяют уравнения (1.118) и (1.119). Масса кислоты m = 10 кг, молярная масса М = 60 г/моль, количество молей кислоты n = 10000/60 = 166,7 моль.
1. Изменение энтропии:
DSТ = DS1 + DS2 + DS3 + DS4 + DS5.
При нагревании твердого вещества от Т1 = 260 до Тпл = 289,8 К
DS1 = mĈртвln(Тпл/Т1) = 10.2039.ln(289,8/260) = 2212 Дж/К.
При плавлении DS2 = nDНпл/Тпл = 166,7.11700/289,8 = 6730 Дж/К.
При нагревании жидкости от Тпл = 289,8 К до Ткип = 391,4 К
DS3 = mĈржln(Ткип/Тпл) = 10.2057.ln(391,4/289,8) = 6182 Дж/К.
При испарении DS4 = nDНисп/Ткип = 166,7.24400/391,4 =
10392 Дж/К.
При нагревании газа от Ткип = 391,4 до Т2 = 430 К
DS5 = mĈргln(Т2/Ткип) = 10.1197. ln(430/391,4) = 1126 Дж/К.
Сумма DSТ = 2212 + 6730 + 6182 + 10392 + 1126 = 26642 Дж/К.
2. Изменение теплоты образования вещества:
DНТ = DН1 + DН2 + DН3 + DН4 + DН5.
Выразим значения теплоемкостей через кДж.кг-1.К-1, значения теплот через кДж.
При нагревании кислоты от Т1 до Тпл
DН1 = mĈртв (Тпл – Т1) = 10.2,039. (289,8 – 260) = 607,6 кДж.
В фазовых переходах:
а) при плавлении DН2 = nDНпл = 166,7.11,700= 1950,4 кДж.
б) при испарении DН4 = nDНисп = 166,7.24,4 = 4067,5 кДж.
При нагревании жидкости от Тпл до Ткип
DН3 = mĈрж (Ткип – Тпл) = 10.2,057. (391,4 – 289,8) = 2090 кДж.
При нагревании газа от Ткип до Т2
DН5 = mĈрг (Т2 – Ткип) = 10.1,197. (430-391,4) = 462 кДж.
Сумма DНТ = 607,6 + 1950,4 + 2090 + 4067,5 + 462 = 9177,5 кДж.
Задача 1.31. Для реакции
2Н2(г) + СО(г) = СН3ОН(г)
вычислить:
а) стандартное изменение энергии Гиббса DrG двумя способами (через DfНо, Sо и через DfGо), изменение внутренней энергии DU и энергии Гельмгольца DrА при Т1 = 298 К; оценить величину полезной работы Wполезн;
б) определить изменение теплоемкости DСр,Т, теплового эффекта реакции DfНТ и энтропии DfSТ при увеличении температуры до Т2 = 400 К;
|
Вещество |
Коэф. в уравн. Ср = а + вТ + сТ2 + с" Т-2 |
|||
В пункте а) вычисляем тепловой эффект реакции DrН298, изменение энтальпии DrS298, энергии Гиббса DrG298, энергии Гельмгольца DrА298.
DrН298 = DfНо(СН3ОН) – (2DfНо(Н2) + DrНоСО) =
201,2 – (2.0 – 110,5) = -90,7 кДж.
DrS298 = Sо(СН3ОН) – (2 Sо(Н2) + SоСО) =
239,8 – (2.130,5 + 197,6) = -218,8 Дж/К.
Расчет энергии Гиббса двумя способами:
1) DrG298 = DrН298 – Т1DrS298 = -90700 – 298(-218,8) = -25498 Дж,
2) DrG298 = DfG(СН3ОН) – (2DfG(Н2)+DfG оСО)=
162400 – (2.0 – 137200) = -25200 Дж.
Результаты расчетов по двум методам одинаковы.
Полезная работа химической реакции равна Wполезн ³ -DrG, где знак равенства соответствует равновесию, а условие “>” вы-полняется для самопроизвольного процесса. Поэтому при Т =
298 К Wполезн > 25200 Дж.
Изменение числа молей газа в ходе реакции:
Dn = n(СН3ОН) – (n (Н2) + n(СО)) = 1 – 2 – 1 = -2.
Изменение внутренней энергии:
DrU298 = DrН298 – DnRТ = -90700 – (-2).8,31.298 = -85747 Дж.
Изменение энергии Гельмгольца:
DrА298 = DrU298 – ТDrS298 = -85747 – 298(-218,8) = -20545 Дж.
В пункте б) определяем изменение теплоемкости DСр,Т, теплового эффекта реакции DНТ и энтропии DSТ при увеличении температуры от Т1 = 298 К до Т2 = 400 К.
Значение DСр,Т находим по уравнению:
DСр = Dа + DвТ + DсТ2 + Dс"Т-2,
где коэффициенты Dа, Dв, Dс и Dс" вычисляем по табличным данным:
Dа = а(СН3ОН) – (2а(Н2) + аСО) = 15,28 – (2.27,28 + 28,41) = -67,69,
Dв = в(СН3ОН) – (2в (Н2) + аСО) = (105,2 – 2.3,26 – 4,1).10-3 =
Dс = с(СН3ОН) = 31,04.10-6,
Dс" = -(2с" (Н2) + с"СО) = -(2. 0,502 + 0,46).105 = -1,464.105.
DСр, 298 = -67,69 + 94,58.10-3.298 + 31,04·10-6.2982 – 1,464.105.298-2 =
38,4 Дж.К-1,
DСр, 400 = -67,69 +94,58.10-3.400 + 31,04·10-6.4002 – 1,464.105.400-2 =
25,81 Дж.К-1.
Изменение теплового эффекта реакции:
DНт = Dа(Т – 298) + (Dв/2)(Т2 – 2982) + (Dс/3)(Т3 – 2983) +
Dс"(1/298 – 1/Т) =
67,69(400 – 298) + 94,58.10-3(4002 – 2982) + 31,04·10-6´
´(4003 – 2983) – 1,464.105(1/298 – 1/400) = + 869,2 Дж.
При вычислении изменения энтропии системы ограничимся первыми тремя членами уравнения:
DSТ = Dа ln(Т2/Т1) + Dв(Т2 – Т1) + (Dс/2)(Т22 – Т12) =
67,69ln(400/298) + 94,58.10-3(400 – 298) +
31,04.10-6(4002 – 2982) = -8,1 Дж.К-1.
В пункте в) определим термодинамические функции DrН400, DrS400, DrG400 и W400 при температуре Т2 = 400 К для прямого направления (образование метанола):
DrН400 = DrН298 + DНт = -90700 + 869 = -89831 Дж,
DrS400 = DrS298 + DSт = -218,8 – 8,1 = -226,9 Дж/К,
DrG400 = DrН400 – Т2DrS400 = -89831 – 400(- 226,9) = + 929 Дж.
Изменение знака DrG с “-” на “+” при увеличении Т от 298 К до 400 К означает, что реакция изменила направление, при
Т = 400 К самопроизвольно протекает обратная реакция – разложение метанола.
Для реакции разложения СН3ОН = 2Н2 + СО при Т = 400 К значение DrG = -929 Дж. Поэтому полезная работа реакции разложения СН3ОН Wполезн. > 929 Дж.
В гл. 9, посвященной энтропии, установлено, что критерием протекания самопроизвольного процесса в изолированной системе является возрастание энтропии. На практике изолированные системы встречаются не часто. Здесь следует сделать одно замечание. Если ограничиться нашей планетой, то она представляет собой достаточно хорошо изолированную систему, и большинство процессов на планете можно рассматривать как протекающими в изолированной системе. Поэтому самопроизвольные процессы идут в сторону возрастания энтропии всей планеты, и именно возрастанием энтропии планеты и характеризуются все самопроизвольные процессы на Земле. Можно, конечно, использовать принцип возрастания энтропии Земли в качестве критерия направленности конкретного рассматриваемого процесса. Однако это очень неудобно, так как придется учитывать энтропию планеты в целом.
На практике чаще имеют дело с закрытыми системами. При анализе самопроизвольных процессов в закрытых системах также можно применить принцип возрастания энтропии.
Рассмотрим реакцию, протекающую в закрытой системе. Закрытая система представляет собой реактор, окруженный термостатом.
Будем полагать, что вся система «реактор + термостат» отделена от окружающей среды изолирующей оболочкой. Как известно, энтропия любой изолированной системы по мере протекания самопроизвольного процесса может только расти. В рассматриваемом случае энтропия представляет собой сумму двух слагаемых - энтропии реакционной системы внутри реактора (А,) и энтропии термостата (А 2). Тогда для изменения энтропии системы в целом можно записать
Предположим, что реакция протекает в условиях постоянного давления и постоянной температуры с выделением теплоты. Постоянство температуры реактора поддерживается хорошей теплопроводностью стенок реактора и большой тепловой емкостью термостата. Тогда теплота, выделяемая в ходе реакции (-АЯ,), поступает из реактора к термостату и
Подставляя величину AS 2 в предыдущее уравнение, получаем
Таким образом, применяя к закрытой системе вместе с термостатом общий принцип возрастания энтропии в изолированной системе при протекания в ней необратимого процесса, получаем простой критерий, который определяет протекание необратимого процесс в закрытой системе (нижний индекс 1 опущен для общности):
В равновесии функция Гиббса закрытой системы достигает минимума, в котором
Выражение (11.25) представляет собой условие равновесия любых закрытых термодинамических систем. Отметим, что стремление системы к равновесию, описываемое уравнением типа (11.24), нельзя объяснять через существование некой «движущей силы». Никаких «движущих сил», аналогичных силам в механике Ньютона, в химических процессах не существует. Химическая система вместе с окружением стремится занять наиболее вероятное состояние из всех возможных, что математически и описывает энтропия полной системы, стремящаяся к максимуму. Таким образом, изотермическое изменение функции Гиббса для закрытой системы, взятое с обратным знаком и поделенное на температуру (-АG/T) - выражение (10.47), представляет собой изменение энтропии полной изолированной системы («термодинамическая система + окружение»), в качестве которой может рассматриваться созданная человеком изолированная система («закрытая система + термостат»), «закрытая система + планета Земля» или «закрытая система + вся Вселенная». Заметим, что во всех обратимых процессах, протекающих при постоянных значениях Тир, изменение фундаментальных функций и энтропии системы вместе с окружением равно нулю на любой стадии процесса.
Итак, в закрытой системе самопроизвольное протекание химического процесса при постоянных значениях температуры и давления, обязательно сопровождается уменьшением функции Гиббса. Реакции, характеризующиеся возрастанием функции Гиббса, самопроизвольно не происходят. Если процесс сопровождается возрастанием функции Гиббса, то его можно осуществить, в большинстве случаев, с совершением работы. Действительно, проведем процесс обратимым путем, но в обратном направлении с уменьшением функции Гиббса. В этом случае будет произведена работа в окружающей среде, которая может быть запасена в виде потенциальной энергии. Если теперь попытаться провести процесс в исходном направлении с возрастанием функции Гиббса, то в обратимом процессе необходимо будет использовать запасенную потенциальную энергию. Следовательно, без совершения работы обратимый процесс, происходящий с возрастанием функции Гиббса, осуществить невозможно.
Тем не менее, можно провести реакцию, в которой происходит возрастание функции Гиббса, и без совершения работы. Но тогда необходимо обеспечить сопряжение невыгодной реакции (AG
> 0) с выгодной (AG

Такие процессы очень часто встречаются в биохимических системах, в которых в роли энергодонорной реакции участвует реакция гидролиза аденозинтрифосфорной кислоты (АТФ). Благодаря сопряжению протекают многие химические и биохимические реакции. Тем не менее, механизм этого сопряжения не столь прост, как это могло бы следовать из вышеприведенной схемы. Отметим, что реакции, в которых участвуют реагенты А и С, независимы. Поэтому протекание реакции С -» D никак не может повлиять на реакцию А -> В. Иногда можно встретить утверждение, что такое сопряжение способно увеличить константы равновесия невыгодных реакций и увеличивать выход продуктов в невыгодных реакциях. Действительно, сложив формально одну реакцию А -» В с некоторым числом (п ) реакций С -> D можно получить сколь угодно большую по величине константу равновесия реакции А + пС -» В + nD . Однако равновесное состояние системы не может зависеть от формы записи химических уравнений, несмотря на суммарное отрицательное изменение функции Гиббса. Поэтому, в сложных системах величины констант равновесия в большинстве случаев не позволяют без проведения расчетов судить о равновесном состоянии. Необходимо иметь в виду, что константы равновесия определяются только структурой участвующих в реакции веществ, и они не зависят от присутствия или реакций других соединений. Простое сложение реакций, несмотря на значительное увеличение констант равновесия, не приводит к увеличению выхода продуктов в равновесной ситуации .
Положение спасает участие в процессе промежуточных продуктов, например,

Но участие промежуточных продуктов не меняет равновесный состав и выход продукта В (предполагается, что равновесное количество промежуточного продукта АС мало). Увеличение количества продукта В можно ожидать только на начальных стадиях процесса, далеких от равновесного состояния благодаря достаточно быстрым реакциям с участием промежуточных продуктов .
Литература
- 1. Степин Б.Д. Применение международной системы единиц физических величин в химии. - М.: Высшая школа, 1990.
- 2. Карапетъянц М.Х. Химическая термодинамика. - М.: Химия, 1975.
- 3. N. Bazhin. The Essence of ATP Coupling. International Scholarly Research Network, ISRN Biochemistry, v. 2012, Article ID 827604, doi: 10.5402/2012/827604
Свободная энергия Гиббса (или просто энергия Гиббса , или потенциал Гиббса , или термодинамический потенциал в узком смысле) - это величина, показывающая изменение энергии в ходе химической реакции и дающая таким образом ответ на вопрос о принципиальной возможности протекания химической реакции.
аправление протекания химической реакции определяет энергия Гиббса (∆G). Еще энергию Гиббса называют изобарно - изотермическим потенциалом. Размерность энергии Гиббса кДж/моль.
При постоянном давлении и температуре (р=const, T=cons)t реакция самопроизвольно протекает в том направлении, которому отвечает убыль энергии Гиббса. Если ∆G < 0, то реакциясамопроизвольно протекает в прямом направлении. Если ∆G > 0, то самопроизвольное протекание процесса в прямом направлении в данных условиях невозможно, а возможно протекание обратного процесса. Если ∆G = 0, то реакция может протекать как в прямом направлении, так и в обратном, и система находится в состоянии равновесия.
Изменение энергии Гиббса в ходе химической реакции (∆ ) не зависит от пути процесса и может быть рассчитано по следствию из закона Гесса: изменение энергии Гиббса в результате химической реакцииравно сумме энергий Гиббса продуктов реакции за вычетом суммы энергий Гиббса исходных веществ с учетом стехиометрических коэффициентов. Например, стандартная энергия Гиббса реакции
aA + bB = сС + dD
где ∆G 0 – стандартная энергия Гиббса образования вещества, кДж/моль.
Энергия Гиббса образования простых веществ равна нулю. ∆ имеет ту же размерность, что и энтальпия, и поэтому обычно выражается в кДж.
Изменение стандартной энергии Гиббса химической реакции может быть также вычислено по уравнению:
∆ = ∆ – Т∆ , где
Т – абсолютная температура,
∆ – изменениеэнтропии.
∆H х.р. – изменениеэнтальпии.
При химическом взаимодействии одновременно изменяется энтальпия, характеризующая теплосодержание системы, и энтропия, характеризующая стремление системы к беспорядку. Уменьшение энтальпии и рост энтропии - две движущих силы любого химического процесса. В состоянии равновесия ∆ =0, значит:
∆ – Т∆ =0 и
Если пренебречь изменениями ∆H 0 х.р. и ∆S 0 х.р с увеличением температуры, то можно определить температуру, при которой устанавливается равновесие химической реакции для стандартного состояния реагентов:
Т равн. =
Многие химические реакции протекают самопроизвольно, т.е. без затрат энергии извне. Одной из движущих сил самопроизвольного химического процесса является уменьшение энтальпии системы, т.е. экзотермический тепловой эффект реакции. Другой – стремление частиц (молекул, ионов, атомов) к хаотическому движению, беспорядку. Мерой хаотичности, неупорядоченности состояния системы служит термодинамическая функция, называемая энтропией (S).
При переходе системы из более упорядоченного состояния в менее упорядоченное состояние (нагревание, испарение, плавление) энтропия возрастает (DS>0). В случае перехода системы из менее упорядоченного состояния в более упорядоченное (охлаждение, конденсация, кристаллизация) энтропия системы уменьшается (DS<0).
В изолированных системах самопроизвольно идут только такие процессы, которые сопровождаются возрастанием энтропии (S>0) – это суть второго закона термодинамики.
Энтропия вещества в стандартном состоянии называется стандартной энтропией (So) и имеет единицу измерения Дж/моль К
Энтропия вещества в газообразном состоянии существенно выше, чем в жидком и твердом состояниях, поэтому об изменении энтропии в химической реакции судят по изменению числа молей газообразных веществ.
Возможность самопроизвольного протекания химического процесса определяется двумя факторами:
Стремлением к образованию прочных связей между частицами, к возникновению более сложных веществ, что сопровождается понижением энергии системы – энтальпийный фактор (DH<0);
Стремлением к разъединению частиц, к беспорядку, что характеризуется возрастанием энтропии – энтропийный фактор (DS>0).
Эти факторы объединяет функция, называемая энергией Гиббса (DG), равная: DG = DH - T DS. (D- это дельта типа, треугольник короче)
Изменение энергии Гиббса служит критерием самопроизвольного протекания химической реакции:
Химическая реакция принципиально возможна, если энергия Гиббса в ходе реакции уменьшается (DG<0);
Химическая реакция не может протекать самопроизвольно, если энергия Гиббса системы возрастает (DG>0), протекает обратная реакция;
Химическая реакция может протекать как в прямом, так и в обратном направлении, т.е. система находится в состоянии равновесия (DG=0).
Из уравнения DG=DH-T DS следует:
Если DН<0 и DS>0, то всегда DG<0, т.е. реакция с выделением теплоты и увеличением степени беспорядка возможна при любых температурах;
Если DH>0 и DS<0, то всегда DG>0, т.е. реакция с поглощением теплоты и увеличением степени порядка невозможна ни при каких условиях;
DH>0, DS<0. Реакция будет протекать в прямом направлении только при условии, что |T DS|>|DH|. Эти реакции протекают при высокой температуре;
DH<0, DS>0. Условие самопроизвольного протекания реакции: |DH|>|T DS|. Такие реакции идут обычно при низких температурах.
Температуру, при которой происходит смена знака энергии Гиббса реакции, можно определить из условия равновесия:
Тр = DH/DS, где Тр – температура, при которой устанавливается равновесие.
Изменение энергии Гиббса системы при образовании 1 моль вещества из простых веществ, устойчивых в стандартных условиях, называется стандартной энергией Гиббса образования вещества (DGof). Стандартная энергия Гиббса образования простых веществ принимается равной нулю.
Стандартную энергию Гиббса химической реакции (DGor) можно рассчитать как сумму стандартных энергий Гиббса образования продуктов реакции за вычетом суммы энергий Гиббса образования исходных веществ с учетом стехиометрических коэффициентов .
Лекция 3
Второй закон термодинамики. Энтропия.
Свойства энтропии. Энтропия – критерий направления самопроизвольного процесса в изолированной системе.
Расчет изменения энтропии при фазовом переходе , нагревании (охлаждении), при протекании химической реакции.
Термодинамические потенциалы и направление самопроизвольного процесса.
Первый закон термодинамики позволяет рассчитать тепловые эффекты различных процессов и работу, совершаемую системой, но ничего не говорит о направлении самопроизвольного протекания процесса.
Второй закон термодинамики устанавливает возможность, направление и предел протекания самопроизвольных процессов. С его помощью можно предсказать направление процесса, не прибегая к дополнительному эксперименту, и определить необходимое изменение условий, позволяющее провести процесс в нужном направлении.
Почему многие экзотермические реакции, сопровождающиеся выделением теплоты, не могут протекать самопроизвольно? И почему все-таки протекают эндотермические процессы, подобные испарению? Почему невозможно построить тепловую машину, КПД которой был бы равен 1? На эти вопросы отвечает II закон термодинамики.
Но прежде, чем перейти к изложению сути II закона термодинамики, необходимо дать характеристику самопроизвольным процессам.
Процессы самопроизвольные и несамопроизвольные,
обратимые и необратимые
Все процессы, протекающие в природе, могут быть разделены на самопроизвольные и несамопроизвольные.
Самопроизвольным, или положительным
, называется процесс, который совершается в системе без вмешательства со стороны окружающей среды. Например, переход теплоты от горячего тела к холодному, плавление льда при t > 0 С.
Свойства самопроизвольных процессов
1) Скорость и движущая сила самопроизвольных процессов измерима (достаточно велика).
2) Самопроизвольные процессы приближают систему к состоянию равновесия, из которого она самопроизвольно выйти не может.
3) Самопроизвольные процессы термодинамически необратимы, т.е. после их протекания систему и окружающую среду одновременно нельзя вернуть в первоначальное состояние: систему можно вернуть в прежнее состояние, затратив работу, но при этом произойдут изменения в окружающей среде (например, изменится энергия окружающих тел).
4) При протекании самопроизвольного процесса совершается работа А н/о (работа необратимого процесса).
Если при осуществлении процесса система может вернуться в исходное состояние, не оставляя видимых изменений в окружающей среде, такой процесс является термодинамически обратимым . Термодинамическое понятие обратимости не совпадает со значением этого термина в химической кинетике. Обратимой в кинетике считают реакцию, результирующая скорость которой определяется разностью скоростей протекания ее в прямом и обратном направлениях, причем на величину этой разности не накладывается каких-либо ограничений.
Для термодинамической обратимости требуется, чтобы реакция проходила в условиях, бесконечно близких к равновесию, когда скорости прямого и обратного процессов различаются на бесконечно малую величину.
Свойства обратимых процессов
1) Обратимые процессы идут с бесконечно малой скоростью через бесконечно большое число стадий, движущая сила их бесконечно мала.
2) При протекании обратимого процесса совершается максимально возможная работа:
А обр. = А max ,
А
обр. > А
н/о
В природе и технике протекают только необратимые процессы. Но любой реальный процесс можно провести в условиях, близких к обратимому процессу. Сравнивая реальный процесс с обратимым, можно в каждом конкретном случае указать пути повышения его эффективности.
Наилучшей моделью обратимого процесса может служить бесконечно медленно протекающий процесс.
Выводы :
1) работа обратимо протекающих процессов максимальна, работа реальных процессов всегда меньше;
2) чем выше степень необратимости процесса, тем меньше работа, производимая системой.
Если I закон термодинамики применим к любым термодинамическим процессам в равной мере, то II закон имеет различное выражение при применении его к обратимым и необратимым процессам.
Формулировки и математическое выражение
II закона термодинамики
Второй закон термодинамики, также как и первый, является эмпирическим. Он не имеет теоретических доказательств и обобщает опытные факты, касающиеся процессов взаимоперехода теплоты и работы. Он имеет несколько формулировок (постулатов), которые эквивалентны и вытекают одна из другой.
Некоторые из формулировок наглядны и непосредственно связаны с опытом, другие более абстрактны, но являются более удобными для математического развития теории. Все они выражают одно и то же содержание, подмечая существование самопроизвольных и несамопроизвольных процессов и различие между ними.
Постулат Клаузиуса : теплота не может сама собой переходить от менее нагретого тела к более нагретому, тогда как обратный переход протекает самопроизвольно .
Постулат Томсона : никакая совокупность процессов не может привести к превращению тепла только в работу, тогда как превращение работы в теплоту может быть единственным результатом процесса .
Постулат Оствальда
: невозможно построить вечный двигатель второго рода, т.е. двигатель, который производил бы работу только за счет поглощения теплоты из окружающей среды без передачи части теплоты теплоприемнику
.
Согласно второму закону термодинамики, даже при обратимом процессе (в котором совершается максимальная работа) в работу может перейти только часть теплоты процесса, другая часть в виде теплоты передается от более нагретых к более холодным частям системы, т.е. КПД всегда меньше единицы. Это явление называется рассеянием (диссипацией) энергии.
Теплота и работа неравноценны . В случае превращения работы в теплоту происходит превращение согласованного, направленного движения микрочастиц системы в беспорядочное, хаотичное движение. Если же происходит превращение теплоты в работу, хаотичное движение должно перейти в направленное. Естественно, что возникновение порядка из беспорядка более затруднительно.
Пример : если ударять молотом по наковальне, то можно обнаружить, что он при этом нагревается, т.е. механическая работа переходит в теплоту. Обратный процесс можно только представить: можно нагреть молот до белого каления, но подпрыгивать от наковальни он не будет.
Вывод: должна существовать какая-то функция состояния системы, характеризующая рассеянную энергию, недоступную для совершения работы.
Эта функция введена Клаузиусом в 1865 г. и названа энтропией S .
Энтропия – это функция состояния, изменение которой равнов приведенной теплоте, сообщенной системе в обратимом процессе:
Выражения (1) являются математической записью II закона термодинамики для обратимых процессов .
В общем виде II закон термодинамики записывается
Знак «=» относится к обратимым процессам, «>» – к необратимым.
Необходимо отметить, что изменение энтропии одинаково как при обратимом, так и при необратимом процессах , но во втором случае происходит большее обесценивание энергии, т.е. переход энергии в состояние, не способное производить работу.
Свойства энтропии. Энтропия – критерий направления
самопроизвольного процесса в изолированной системе
1. Энтропия – функция состояния системы , т.е. ее изменение S зависит только от энтропии исходного и конечного состояний системы.
2. Энтропия характеризует вероятность реализации системы. Чем больше энтропия, тем больше способов реализации системы. Например, энтропия возрастает при распаде молекул ВМС на отдельные фрагменты, при переходе вещества из твердого в жидкое и газообразное состояние при постоянной температуре, при нагревании вещества (так как усиливается тепловое движение молекул и возрастает беспорядок). Количественно эта взаимосвязь выражается формулой Больцмана
S = k lnW ,
Где W – термодинамическая вероятность; k – константа Больцмана, k = 1,3810 -23 Дж/К.
Термодинамическая вероятность
W
– это число микросостояний системы, с помощью которых реализуется данное макросостояние. Макросостояние системы характеризуется параметрами состояния (p
, V
, T
, хим. состав). Но термодинамическая система состоит из огромного числа микрочастиц, имеющих определенную энергию, скорость, направление движения, поскольку находятся в непрерывном хаотичном движении. При равновесии макросостояние не изменяется, т.е. макросвойства (p
, V
, T
, хим. состав) остаются постоянными, а микросвойства (положение частицы в объеме системы, энергия, скорость ее) непрерывно меняются. Наблюдаемое макросостояние осуществляется разными микросостояниями, количество которых характеризует термодинамическая вероятность. В отличие от математической вероятности, равной отношению числа благоприятных событий к общему числу возможных событий, а поэтому всегда меньшей единицы, термодинамическая вероятность может быть очень большой величиной.
нагревании (охлаждении), при протекании химической реакции
Для реальных (необратимых) процессов II закон термодинамики записывается в идее неравенства, что затрудняет расчет изменения энтропии S при их протекании. Но энтропия – функция состояния системы, и ее изменение не зависит от пути проведения процесса. Поэтому для расчета S при протекании различных процессов воспользуемся уравнением II закона для обратимых процессов:
Изменение энтропии при фазовых превращениях
Фазовое превращение (фазовый переход) – процесс, связанный с изменением агрегатного состояния вещества.
Характерной особенностью этих процессов является то, что они протекают при постоянной температуре – температуре фазового перехода Т ф.п. .
Тогда, согласно II закону термодинамики
Где Q ф.п . – тепловой эффект фазового перехода.
При р = const теплота равна изменению энтальпии:
Изменение энтропии при нагревании (охлаждении).
Применим уравнение (1) к изобарному процессу (р = const ).
Где С р – молярная изобарная теплоемкость вещества, Дж/(мольK).
Проинтегрируем последнее уравнение в определенных пределах (при изменении температуры от Т 1 до Т 2):
Если в исследуемом температурном интервале теплоемкость мало зависит от температуры, т.е. можно использовать среднюю изобарную теплоемкость  , то решение уравнения (2) имеет вид:
, то решение уравнения (2) имеет вид:
Изменение энтропии в ходе химической реакции (при Т = const ).
Так как энтропия – функция состояния, то ее изменение в ходе химической реакции можно рассчитать по уравнению:
Где S j , S i – энтропии продуктов реакции и исходных веществ соответственно при температуре реакции; j , i – стехиометрические коэффициенты.
В справочной литературе приведены стандартные энтропии веществ при температуре 298 К.
Если реакция протекает при температуре, отличной от 298 К, то энтропию вещества рассчитывают по уравнению (3), приняв для удобства за Т 1 температуру 298, а за Т 2 – температуру реакции:
Где S – изменение энтропии вещества при нагревании (или охлаждении) вещества от 298 К до температуры Т .
После подстановки (5) в формулу (4) для каждого участника реакции получим формулу для расчета изменения энтропии в ходе реакции, протекающей при температуре Т :
II закон термодинамики:
 или
или  .
.
Объединенное математическое выражение:
Рассмотрим соответствующие процессы.
1. Изобарно-изотермический процесс (p , T = const ).
Уравнение (6) запишется (из математики: любую константу можно внести под знак дифференциала, а сумма дифференциалов равна дифференциалу от суммы )
Где H – TS = G –свободная энергия Гиббса. При p , T = const изменение энергии Гиббса связано с выполнением полезной работы:
В обратимо протекающем процессе  . Тогда
. Тогда
Таким образом , свободная энергия Гиббса является изобарно-изотермическим потенциалом , так как ее уменьшение характеризует максимальную работу этого процесса.
Если единственным видом работы является работа расширения (сжатия), т.е.  , то в необратимом, а, следовательно, самопроизвольно протекающем процессе
, то в необратимом, а, следовательно, самопроизвольно протекающем процессе
Неравенства (7), (8) являются условием самопроизвольного протекания процесса при постоянстве соответствующих параметров : самопроизвольно могут протекать только те процессы, которые приводят к понижению свободной энергии системы; система приходит в состояние равновесия, когда свободная энергия достигает минимального значения.
Критерии оценки направления самопроизвольного протекания процессов
| Критерий Оценки | Ограничения | Условие Равновесия | Условие протекания самопроизвольного процесса |
| Энтропия S | Изолированная Система |
ΔS = 0, S = S
max | ΔS > 0 |
| Изобарно-изотермический потенциал G | р,Т = const | ΔG = 0, G
= G
min | ΔG |
| Изохорно-изотермический потенциал F | V, Т = const | ΔF = 0, F = F
min | ΔF |
Изменение соответствующего термодинамического потенциала в ходе какого-либо процесса, протекающего при температуре Т , вычисляют по уравнению Гиббса-Гельмгольца:
– для изобарно-изотермического процесса
,
G
T
=
H
T
–
T
S
T
(9)
– для изохорно-изотермического процесса
.
F
T
=
U
T
–
T
S
T
(10)
H
и
U
– изменение полной энергии в системе при р =
const
и V
=
const
соответственно; G
и
F
– энергия, которая связана с производством полезной работы; T
S
– энергия, которая перешла в энергию хаотичного (теплового) движения частиц, вследствие чего она уже не может перейти в работу. Поэтому G
и
F
называют еще свободной энергией, а T
S
– связанной энергией.
Легко установить связь между G
и F
. Вычитая из уравнения (9) уравнение (10), и учитывая, что U
= Н –
nRT
, получим:
Термодинамические потенциалы могут играть роль характеристических функций. Это значит, что с помощью их производных можно выразить свойства системы, необходимые для ее характеристики.
Например, изобарно-изотермический потенциал является функцией двух параметров – давления и температуры, т.е.
Запишем dG в виде суммы частных производных
Получаем:

Многие процессы протекают без подвода энергии от внешнего источника. Такие процессы называют самопроизвольными .
Примерами самопроизвольных процессов могут служить падение камня с высоты, течение воды под уклон, переход теплоты от более нагретого тела к менее нагретому.
Человеческий опыт показал, что самопроизвольные процессы в обратном направлении не могут протекать самопроизвольно, т.е. самопроизвольно не потечет вода в гору, камень не полетит вверх, а теплота не перейдет от холодного тела к нагретому.
(хотя с точки зрения первого закона термодинамики, одинаково правдоподобны как процесс перехода тепла от горячего тела к холодному, так и обратный процесс, т.е. переход от тепла от холодного тела к горячему, ибо и в том и в другом случаях соблюдается закон сохранения и превращения энергии)
Многие химические реакции также протекают самопроизвольно, например , образование ржавчины на металлах, реакция натрия с водой, растворение соли в воде и др.
Чтобы понимать химические процессы и управлять ими, необходимо знать ответ на вопрос: каковы движущие силы и критерии самопроизвольных процессов?
Одной из движущих сил химической реакции является рассмотренное нами ранее уменьшение энтальпии системы, т.е. экзотермический тепловой эффект реакц ии.
Как показывает опыт, большинство экзотермических реакций (?Н <0) протекают самопроизвольно. – Почему?
Однако условие?Н <0 не может быть критерием! Самопроизвольного течения реакций, так как существуют самопроизвольные эндотермические химические реакции, у которых?Н >0, например, взаимодействие метана с водяным паром при высокой температуре.
Следовательно, кроме уменьшение энтальпии системы (энтальпийного фактора) имеется другая движущая сила самопроизвольного процесса.
Такой силой является стремление частиц (молекул, ионов, атомов) к хаотичному движению, а системы – к переходу от более упорядоченного состояния к менее упорядоченному.
Например, представим пространство, в которое помещено вещество, в виде шахматной доски, а само вещество – в виде зерен. Каждая клетка доски соответствует определенному положению и уровню энергии частиц. Если частицы распределяются по всему пространству, то вещество находится в газовом состоянии; если частицы займут только небольшую часть пространства, то вещество перейдет в конденсированное состояние. Все высыпанные зерна распределяются на доске более или менее равномерно. На каждой клетке доски окажется определенное число зерен. Положение зерен после каждого рассыпания соответствует микросостоянию системы, которое можно определить как мгновенный снимок, фиксирующий расположение частиц в пространстве. Каждый раз мы получаем систему в одном и том же макросостоянии. Число подобных микросостояний, удовлетворяющих ожидаемому макросостоянию (при достаточно большом количестве частиц) очень велико.
Например , коробка с ячейками, в которой находятся шары: так в 9 ячейках находятся 4 шара – это модель макросистемы . Шары по ячейкам можно разложить 126 различными способами, каждый из которых является микросостоянием.
Число микросостояний, посредством которых реализуется данное макро состояние, связано с термодинамической вероятностью W . Энтропия определяется термодинамической вероятностью : она тем выше, чем больше способов реализации макросостояния .
Поэтому считают, что энтропия – мера неупорядоченности системы.
Математически связь энтропии с числом микросостояний установил Л. Больцман в конце 19 века, выразив ее уравнением:
S = k * ln W ,
где W - термодинамическая вероятность данного состояния системы при определенном запасе внутренней энергии U и объеме V ;
k – постоянная Больцмана, равная 1,38*10 -23 Дж/К.
Пример с шарами, конечно, очень нагляден, но он коварен, так как на основании его интуитивно под упорядоченностью системы иногда понимают расположение частиц в пространстве .
Однако, в действительности под термодинамическим состоянием подразумевается, главным образом, расположение частиц (например, молекул) по возможным уровням энергии (каждый вид движения –колебательное, вращательное, поступательное- характеризуется своим уровнем энергии).
Энтропия также зависит от массы частиц и их геометрического строения.
Кристаллы имеют
наименьшую энтропию (так их частицы могут колебаться только около некоторого
состояния равновесия), а газы – наибольшую, так как для их частиц возможны все
три вида движения. S T
Всякому веществу можно приписать определенное абсолютное значение энтропии.
Конечно, энтропии веществ обычно не рассчитывают на основании уравнения Больцмана. Их определяют по уравнению классической термодинамики с учетом теплоемкости данного вещества и теплот фазовых переходов.
Значение энтропии различных веществ при 298 К и давлении 1 атм. (S 0 298) являются табличными данными.
На основании данных о стандартной энтропии веществ можно рассчитать изменение энтропии различных химических процессов. Поскольку энтропия является функцией состояния , то ее изменение не зависит от пути процесса и равно разности энтропий продуктов реакций и исходных веществ:
?S 0 реакц .= ? ? i S 0 - ? ? jS 0
Во многих случаях изменение энтропии процесса можно оценить качественно:
· Так, энтропия всегда увеличивается при переходе из конденсированного состояния (твердого или жидкого) в парообразное.
· Энтропия всегда возрастает при растворении твердого или жидкого вещества, причем, чем больше степень диссоциации, тем заметнее увеличивается энтропия. При растворении газов, напротив, энтропия уменьшается.
· Чем сложнее состав вещества, тем больше энтропия. Например, для оксидов марганца МnO , Mn 2 O 3, Mn 3 O 4 энтропия равна соответственно 61,50; 110,5; 154,8 кДЖ/моль*К.
· В химических реакциях энтропия возрастает, если в результате их увеличивается количество газообразных веществ. Например, в реакции термического разложения карбоната кальция:
СаСО 3(т) = СаО (т) + СО 2(г)
Второе начало (закон) термодинамики регламентирует принципиальную возможность протекания различных процессов. В середине 19 века этот закон был сформулирован в виде нескольких постулатов. Наиболее известные из них следующие:
· Невозможно осуществить перенос тепла от более холодного тела к более горячему, не затрачивая на это работу .
(Р. Клаузиус)
и с использованием понятия энтропии:
· В изолированных системах самопроизвольно идут процессы, при которых происходит увеличение энтропии. (? S изолир. >0)
Всякая изолированная система самопроизвольно стремиться принять состояние, характеризующееся максимальной термодинамической вероятностью.
На основании уравнения Больцмана можно показать, что любой необратимый процесс, самопроизвольно протекающий в изолированной системе, характеризуется увеличением энтропии. Пусть в изолированной системе находятся два химически не взаимодействующих газа, например гелий и неон, при одинаковых условиях, разделенные перегородкой. В этом состоянии термодинамическая вероятность системы w 1 . При удалении перегородки газы начинают самопроизвольно диффундировать друг в друга до тех пор, пока молекулы каждого газа равномерно не распределятся по всему объему. В конечном состоянии термодинамическая вероятность w 2 . Система самопроизвольно перешла из менее вероятного состояния в более вероятное (w 2 > w 1). Энергетический обмен системы с внешней средой отсутствует, следовательно, единственная причина протекания этого процесса - увеличение энтропии.
Другими словами, процессы протекают самопроизвольно лишь в сторону менее упорядоченного состояния, т.е. нарастания беспорядка. Именно поэтому испарение жидкости, растворение соли в воде или смешение газов происходит самопроизвольно, а вместе с тем обратные процессы без обмена энергией с окружающей средой невозможны.
Следовательно, увеличение энтропии является критерием самопроизвольного протекания процессов только в изолированных системах, т.е. не обменивающихся энергией с внешней средой, а это довольно редкий случай. В открытых и замкнутых системах, кроме изменения энтропии, на направление процесса влияет еще и изменение энтальпии.
Вопрос 5. Энергия Гиббса и Гельмгольца. Критерий самопроизвольного протекания процессов.
Какие же процессы идут самопроизвольно в неизолированных системах? При взаимодействии водорода с кислородом самопроизвольно образуется вода:
2Н 2(г) +О 2(г) = 2Н 2 О (г)
В этой реакции энтропия уменьшается, но выделяется большое количество теплоты (? S <0, ?Н <0), т.е. самопроизвольному протеканию процесса способствует уменьшение энтальпии.
Самопроизвольно происходит и растворение хлорида аммония в воде:
NH 4 Cl (тв) + aq = NH 4 + (р) + Cl - (р)
Этот процесс сопровождается понижением температуры (поглощение теплоты) и увеличением энтропии (? S > 0, ?Н > 0), причем главную роль играет последний фактор.
В термодинамике вводится новая функция, связывающая две предыдущие величины – энергия Гиббса.(G )
G = H – TS
Основная ценность этой функции заключается в том, что ее изменение при постоянной температуре и давлении определяет самопроизвольность процессов.
? G = ( ? H – T ? S ) <0
· В классической термодинамике под энтропией понимают такое свойство системы, изменение которого при обратимом процессе численно равно отношению теплоты к температуре протекания процесса:
? S = Q/T ; T ? S=Q
· В термодинамике обратимым называют такой процесс, который проводится бесконечно медленно и так, чтобы система находилась все время практически в состоянии равновесия.
Таким образом, величина ? G характеризует ту часть изменения внутренней энергии, которая может быть превращена в полезную работу.
При условии постоянства объема пользуются термодинамической функцией, которая называется Энергией Гельмгольца (F ):
F = U – T ? S
В изохорном процессе полезная работа определяется изменением энергии Гельмгольца, а условием самопроизвольности процесса является ее уменьшение ? F <0.
В химии обычно пользуются энергией Гиббса, поскольку чаще всего химические реакции проводят при постоянном (атмосферном) давлении.
Итак, в неизолированной системе процесс преимущественно происходит самопроизвольно, если ему соответствует уменьшение энергии Гиббса. (? G <0.)
При ? G =0 состояние системы соответствует равновесию.
При ? G > 0 -процесс преимущественно не протекает в прямом направлении
Анализ уравнения ? G =( ? H – T ? S ) показывает, что знак величины ? G , а значит, термодинамическая возможность самопроизвольного протекания реакции зависят от двух факторов: энтальпийного (энергетического) и энтропийного . С одной стороны, система стремится занять прийти к минимальному уровню энергии, выделив часть ее в виде теплоты или работы (? H <0). С другой стороны, система стремится занять наиболее вероятное состояние, характеризующееся максимумом молекулярного беспорядка, т.е. максимумом энтропии (? S >0). В этом случае энтальпийный и энтропийный факторы действуют в направлении, благоприятствующему протеканию реакции.
Рассмотрим варианты:
а) ? H <0; ? S >0; в этом случае? G <0 при всех значениях температуры, процесс термодинамически возможен при любой температуре.
б) ? H <0; ? S <0; в этом случае? G <0 при Т< , т.е. при реакция термодинамически возможна при при сравнительно низкотемпературном режиме;
в) ? H >0; ? S >0; в этом случае? G <0 при Т> , процесс возможен при высоких температурах;
г) ? H >0; ? S <0; в этом случае? G <0 - оба фактора действуют в неблагоприятном направлении, реакция термодинамически невозможна при любых значениях температур.
Первый способ расчета аналогичен методу оценки изменения энтальпии реакции по табулированным энтальпиям образования различных веществ. В таблицах сведены и величины ? G 0 обр.298 и точно также принято, что для простых веществ ? G 0 обр.298 =0
? G 0 реакц. = ?? i ? G 0 обр.прод. - ?? j ? G 0 обр.исх.
i j
Второй способ основан на расчете сначала величин ? H реакц.. и ? S реакц. для данного процесса, а потом исходя из них – величины ? G 0 реакц по формуле:
? G 0 реакц = ? H 0 реакц. – 298 ? S 0 реакц.
Данный способ хорош тем, что позволяет оценить, как изменится знак ? G 0 реакц при изменении температуры.
Хотя энтальпия и энтропия веществ зависят от температуры, но для реакции изменение этих величин незначительно, поэтому приближенно считают, что в некотором интервале температур ? H реакц.. и ? S реакц величины практически постоянные.
Для простых веществ, находящихся в термодинамически устойчивых состояниях ? G 0 =0.